
What Are KCNQ2-Related Disorders
A spectrum of genetic neurological conditions caused by mutations in the KCNQ2 gene, primarily resulting in neonatal-onset epilepsy
What Are KCNQ2-Related Disorders
A spectrum of genetic neurological conditions caused by mutations in the KCNQ2 gene, primarily resulting in neonatal-onset epilepsy
KCNQ2-Related Disorders
Discovery of the KCNQ2 Gene
 The history of the identification of KCNQ2 developmental and epileptic encephalopathyas an identifiable disorder begins with the identification and characterization of another related disorder, benign familial neonatal epilepsy (BFNE). This condition was initially described as a syndrome in 1964 by doctors Rett and Teubel. They reported a family with eight affected individuals over 3 generations. The youngest infant had the onset of seizures at 3 days of age described as tonic-clonic events occurring multiple times per day. The EEGs were normal in between seizures and children developed appropriately after the seizures stopped. This typically happened later in infancy. Over the next twenty years, additional families with similar stories were described. In a few instances, seizures persisted into later life but the outcomes were otherwise favorable. The pattern of inheritance was determined to be autosomal dominant (see the Affected Populations section for further explanation) and genetic testing linked the disorder to the long arm of chromosome 20 (see the Cause section for further definition). In 1998, researchers identified a gene in the region that appeared similar in structure to a potassium channel within the heart. This new gene was named, according to convention, KCNQ2. Subsequently, several families were identified in which the outcomes were not benign, having either persistent seizures that did not respond to medication, developmental impairment, or both. This prompted a group of researchers to screen patients with severe neonatal epilepsy syndromes for mutations in KCNQ2. Eight cases were identified from that group of 80 patients, with those children sharing many characteristics. Since that initial paper in 2011, many more individuals have been diagnosed and the syndrome has been defined further.
The history of the identification of KCNQ2 developmental and epileptic encephalopathyas an identifiable disorder begins with the identification and characterization of another related disorder, benign familial neonatal epilepsy (BFNE). This condition was initially described as a syndrome in 1964 by doctors Rett and Teubel. They reported a family with eight affected individuals over 3 generations. The youngest infant had the onset of seizures at 3 days of age described as tonic-clonic events occurring multiple times per day. The EEGs were normal in between seizures and children developed appropriately after the seizures stopped. This typically happened later in infancy. Over the next twenty years, additional families with similar stories were described. In a few instances, seizures persisted into later life but the outcomes were otherwise favorable. The pattern of inheritance was determined to be autosomal dominant (see the Affected Populations section for further explanation) and genetic testing linked the disorder to the long arm of chromosome 20 (see the Cause section for further definition). In 1998, researchers identified a gene in the region that appeared similar in structure to a potassium channel within the heart. This new gene was named, according to convention, KCNQ2. Subsequently, several families were identified in which the outcomes were not benign, having either persistent seizures that did not respond to medication, developmental impairment, or both. This prompted a group of researchers to screen patients with severe neonatal epilepsy syndromes for mutations in KCNQ2. Eight cases were identified from that group of 80 patients, with those children sharing many characteristics. Since that initial paper in 2011, many more individuals have been diagnosed and the syndrome has been defined further.
What are KCNQ2-related epilepsies?
Mutations in the KCNQ2 gene are responsible for a spectrum of neonatal-onset epilepsy syndromes, encompassing both severe early-onset epilepsies known as developmental and epileptic encephalopathies (DEEs), as well as milder forms referred to as self-limited (benign) neonatal epilepsies. The impact of these genetic variations can vary widely, leading to diverse symptoms and varying degrees of disorder severity based on the specific type of KCNQ2-related epilepsy an individual has.
It is important to note that KCNQ2 itself is not a medical condition; rather, it is the name of the gene that undergoes pathogenic alterations. When epilepsy can be attributed to a disease-causing variant in the KCNQ2 gene, it is categorized as a KCNQ2-related disorder. This distinction helps clarify the underlying genetic basis of the condition and facilitates accurate diagnosis and management strategies.
Phenotypes and Symptoms
KCNQ2-related disorders encompass a range of neonatal epileptic conditions that can vary in severity. These conditions include self-limited familial neonatal epilepsy (SLFNE) at the milder end and neonatal-onset developmental and epileptic encephalopathy (DEE) at the more severe end.
- KCNQ2-SLFNE, seizures typically occur in otherwise healthy infants between two and eight days after birth, but they disappear on their own between the first and twelfth month of life. There is a period without seizures between birth and the onset. These seizures often involve sudden movements, with affected limbs becoming stiff, and can be accompanied by temporary pauses in breathing and bluish discoloration. Video EEG recordings help identify these seizures as starting in a specific area of the brain and involving muscle stiffness, with some movement during each seizure. It's important to note that about 30% of individuals with KCNQ2-SLFNE may develop epileptic seizures later in life.
- KCNQ2-DEE is characterized by multiple seizures occurring daily, starting within the first week of life. These seizures are often characterized by muscle stiffness and can involve specific body parts. The seizures typically stop between nine months and four years of age. EEG recordings at the beginning may show patterns of brain activity called burst suppression or abnormal activity in multiple areas. Early brain MRI scans may reveal increased density in certain brain regions, and later scans may show loss of brain tissue or changes in white matter. Individuals with KCNQ2-DEE usually experience moderate to severe developmental impairments.
It's important to understand that KCNQ2-related disorders represent a spectrum of conditions, with varying symptomatology and outcomes. These disorders require careful medical evaluation and management to ensure appropriate support and treatment for affected individuals.
Additional phenotypes:
- Neonatal encephalopathy with non-epileptic myoclonus
- Profound encephalopathy at birth with hypotonia and poor respiratory effort
- Spontaneous episodes of jerking movements (myoclonus); may also be provoked by touch and/or attempted arousal
- Defining neonatal video EEG findings: invariant burst-suppression EEG background; body and limb jerks (myoclonus) without cortical accompaniment
- Epileptic spasms and other seizure types and profound encephalopathy emerging in early infancy
- Progressive volume loss and hypomyelination on brain MRI
- Non-neonatal-onset developmental and epileptic encephalopathy
- Infantile- or childhood-onset epilepsy, including West syndrome
- Neurodevelopment is usually impaired from birth, although normal early development with stagnation at onset of seizures has been described.
- Subsequent developmental impairment is moderate to severe, even if seizures are controlled.
- Brain MRI may show mild volume loss.
- Isolated intellectual disability (ID)
- ID without epilepsy
- Motor and speech delays may be present.
- Brain MRI is normal or shows mild myelinization delay.
KCNQ2 and Seizures

Seizures are one of the hallmarks of KCNQ2. Nearly all of those affected by KCNQ2 experience seizures in the first several days of life. This is most often the symptom leading to testing resulting in a diagnosis of KCNQ2. Following this early, neonatal period, there is wide variability in the seizure activity each patient experiences. Many patients are able to gain good control of seizures with available medications, while some have refractory seizures which are difficult to control and continue into later life. Some see seizures dissipate early following initial seizure activity. Even for those whose seizures resolve during early life, many continue to be at risk for febrile or sporadic seizure activity even as they are older.
There are various seizure types experienced by those with KCNQ2. The classifications of seizures used by experts have recently been updated, and are described in the attached image.
KCNQ2 and Autism
Many children diagnosed with KCNQ2 also display symptoms of autism, as a result of the impact of KCNQ2. In addition to the general cognitive and developmental disabilities affecting nearly all of those with KCNQ2, many children also display repetitive movements, poor eye contact, self-harm, sensitivity to sound, or other symptoms associated with autism. As such, therapies known to be effective for the treatment of autism may be helpful. SimmonsVIP, one of the leaders in genetic research related to autism, is researching KCNQ2 as one of the genetic causes of autism.
Diagnosing KCNQ2
If seizures occur shortly after birth or if there is a family history of seizures, KCNQ2-related epilepsy may be suspected.
Clinical Testing and Work-up Evaluations
EEG: One of the first steps in the evaluation of seizures is to characterize the patterns of brain activity associated with the seizures. This is done by performing an electroencephalogram or EEG. This is a painless and non-invasive means of recording the patterns of electrical activity of the brain. Electrodes placed on the scalp pick up and record the electrical waves during periods of activity, sleep, and during seizures. KCNQ2 is often associated with a burst-suppression pattern on EEG but may have other non-specific abnormalities and the EEG is typically not normal between seizures, in contrast to SLFNE, in which the EEG can normalize.
When seizures present in infancy, there are a number of potential causes that physicians and insurers may need to excluded before genetic testing is pursued. This often depends on the presentation and other clinical factors. Tests that may be performed include evaluations for infection, electrolyte disturbance, metabolic disorders, and structural problems in the brain.
MRI: Magnetic Resonance Imaging (MRI) is a radiological technique that produces detailed images of cross-sections or slices of the brain by using a magnetic field. The images can provide information concerning any malformation of the brain structures or other types of lesions commonly seen in epilepsy. The malformations of the potassium channel caused by the KCNQ2 mutation, are too small to be detected on an MRI.
Genetic Testing: The diagnosis of KCNQ2 is ultimately made by molecular genetic testing. This can be done by examining only the potassium channel gene or by a genetic test that looks for mutations in a number of genes associated with epilepsy in infancy, or even whole genome or whole exome sequencing, which screen all, or nearly all genes.
Treatment and Therapies
Treatment decisions may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, developmental pediatricians, and/or other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
In some cases, it is possible that treatment with anticonvulsant drugs may help reduce or control various types of seizure activity associated with KCNQ2. Anticonvulsant medications have many different mechanisms of action and it is not entirely clear which medications are best for KCNQ2. Some reports suggest that children respond best to medications that affect how sodium or potassium ions flow into nerve cells; however, the number of children evaluated in these studies may be too small to draw these conclusions. In practice, seizures are treated with a wide range of different medications, most often in combinations, in children with KCNQ2. If seizures fail to respond to medication, other treatments including specialized diets, devices, and surgeries may be considered.
In some cases, implantable devices like vagus nerve stimulation (VNS) or responsive neurostimulation (RNS) may be considered when medications are not helpful.
Genetics in KCNQ2
KCNQ2 is caused by a mutation on the KCNQ2 gene, located on chromosome 20.
Chromosomes: Chromosomes are located in the nucleus of human cells and carry the genetic information for each individual. Human body cells normally have 46 chromosomes in each cell. Pairs of human chromosomes numbered from 1 through 22 are called autosomes, in addition to the sex chromosomes, which are designated X and Y (males have one X and one Y chromosome and females have two X chromosomes). Each chromosome has a short arm designated “p” and a long arm designated “q”. Each chromosome is further subdivided into many bands that are numbered. For example, “chromosome 11p13” refers to band 13 on the short arm (p) of chromosome 11. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
Genes: Each chromosome contains thousands of genes, and each of those genes contains the code with the “instructions” for all of the components of the human body. Each gene is a segment of DNA with instructions for a particular component. In the case of KCNQ2, there is an error in the code in the KCNQ2 gene. This error may be inherited from a parent, or occur spontaneously. In the cases in which the mutation is inherited, only one parent may carry the mutation for it to be seen in the child. In some cases, the parent may not have ever displayed symptoms of the disease, if the mutation is only on a small portion of the parent’s cells, but the child may have the mutation in many more cells, leading to symptomatic disease. In many cases, the mutation in KCNQ2 is not inherited, and is called “de novo”. De novo mutations in the KCNQ2 gene occur when the genes are copied over and over as cells are dividing soon after conception. As the genes are copied, sometimes there is a random error in the genetic code, like a “typo”. This error can be a deletion of one of the “letters” in the code or can be a substitution of an incorrect “letter” in the code.
Nucleotides: Genes are made up of nucleotides. The genes carry the code for making the human body using sequences of nucleotides. There are approximately 3 billion nucleotides comprising a human’s DNA. In most cases of KCNQ2, there is an error in just one of these 3 billion nucleotides, but it is in a location that codes for a critical protein.
There are only four nucleotides used in all genetic codes; they are cytosine (C), thymine (T), adenine (A), and guanine (G). Various combinations of three nucleotides, depending on which nucleotides and in what order, code for various amino acids. These amino acids, in turn, are the building blocks of proteins. It is in this way, that the nucleotides that make up the code for the genes, determine what proteins are formed. When there is an error or mutation in the sequence of nucleotides the resulting protein is malformed. In the case of KCNQ2, the mutation (the error in which nucleotide is present) is in the DNA sequence coding for KCNQ2, or the potassium channel. Because the potassium channel is important for the brain sending signals throughout the body, even a very minor change in the structure of the protein which results from just a single nucleotide change, can have a significant impact.
Gain or Loss of Function Mutations: Depending on where the error is, and what the error is (which nucleotide is replaced or missing), the mutation can result in the potassium channel having a “loss of function” LOF (being closed more than it should be) or having a “gain of function” GOF (being open more than it should be). The vast majority of KCNQ2 cases are due to a loss of function in the potassium channel. With a growing community of patients being diagnosed and data on the variants being collected, there is an increasing understanding of which mutations, or variants, cause a gain of function and which cause a loss of function. Understanding whether a patient is gain of function or loss of function can have implications for a recommended treatment course, particularly as more targeted therapies are being developed.
Reading Your Genetic Report
Most families receive the news of their child’s KCNQ2 diagnosis verbally from a medical professional, often followed by a written report from the lab performing the testing. This lab report will contain information about the specific variant of KCNQ2 mutation that may be useful in identifying potential treatments or possible outcomes.
To explain what the information means, we will use a specific example from a patient’s GeneDx genetic report. The report says:
p.Arg333Trp
(CGG>TGG)
c.997C>T
The report is written in a “top-down” approach. Starting with the first line describing the amino acid (or protein building block that is produced), the second line describing what the sequence of nucleotides is, that caused those particular amino acids to be produced, and the last line describing the coding DNA, which codes for the nucleotides. However, in actuality, the process happens in reverse order, as described in the section Genetics in KCNQ2. The mutation comes into play with an error in a nucleotide in the coding DNA, resulting in the error in the nucleotide sequence, resulting in an error in the amino acid produced.
The first line: p.Arg333Trp tells about the protein that is coded for (the “p” is an abbreviation for protein). In the case of this particular variant, the mutation results in the production of the amino acid tryptophan (abbreviated Trp) in place of arginine (abbreviated Arg) at location 333. The abbreviation is written:
- p. for protein
- correct amino acid, in this case, Arg or arginine
- location of the mutation, in this case, 333
- actual amino acid, in this case, Trp or tryptophan
The second line (CGG>TGG) means that this patient has the sequence of nucleotides TGG in place of CGG (which is the normal sequence). The sequence TGG codes for tryptophan, which explains why he has that amino acid instead of arginine (which is coded for by CGG). The “>” sign means it is a substitution. If there is a “_” sign it means it is a deletion. The table shows how these three-nucleotide sequences, or “DNA Codons”, code for the amino acids.
The last line c.997C>T explains the mutation in the coding DNA (the coding DNA is what codes for the protein, described in the first line). This report says that the nucleotide cytosine (abbreviated as C) is replaced with thymine (abbreviated as T) at the nucleotide number (or position) 997. That’s why the letters are TGG instead of CGG in the sequence shown in line 2, above.
CAUSE
The KCNQ2 Gene
The gene that is altered in patients with KCNQ2 developmental and epileptic encephalopathy (KCNQ2) is the gene for a potassium channel within the brain, located on chromosome 20q13.33.
The KCNQ2 gene belongs to a family of other ion channel genes and is sometimes abbreviated Kv7.2. Ion channels are pores in the cell membrane, around the outside of the cells, with gates that allow charged atoms (ions) to flow into and out of cells. These ions play a key role in a cell’s ability to generate and transmit electrical signals.
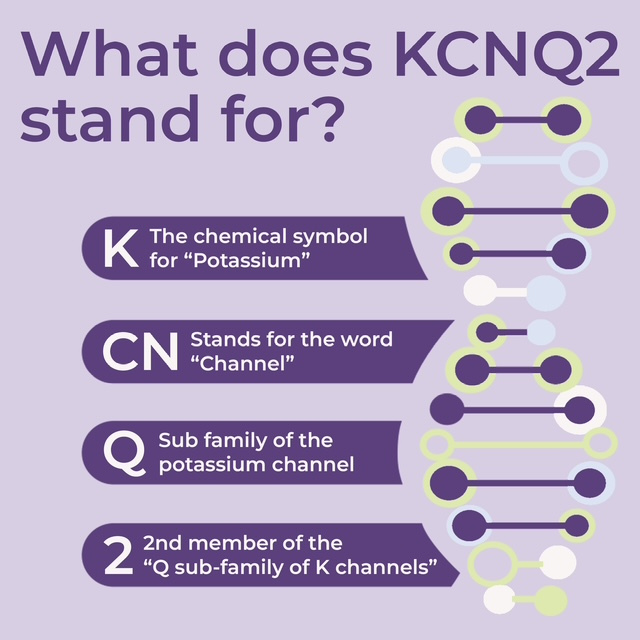
The genes for the ion channels share important properties and are named to reflect them. “K” is the chemical symbol for potassium, which is a positively charged ion. CN is an abbreviation for the channel. The KCNQ2 gene is the 2nd member of the Q subfamily, which indicates that the channel is voltage-gated. Being voltage-gated means that the channel opens and closes according to the charge in environment in the cell. Mutations in the KCNQ2 gene cause a spectrum of disease that ranges from benign seizures in infancy to developmental and epileptic encephalopathy. These differences are likely based on the degree of dysfunction in the potassium channel. Those mutations that cause encephalopathy are typically located in one of several particular areas. However, recent literature suggests that distinguishing presentations may be more complex than initially thought; other factors, other than the location of the KCNQ2 mutation, affect outcomes, which can vary significantly even for individuals with the same variant of the mutation.
KCNQ2 is an autosomal dominant disorder. Most genetic diseases are determined by the status of the two copies of a gene, one received from the father and one from the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease.
The abnormal gene can be inherited from either parent or can be the result of a new mutation (gene change, or de novo mutation) in the affected individual. If one of the parents carries the gene, the risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females. In some individuals, the disorder is due to a new (de novo) genetic mutation that occurs in the egg or sperm cell. In such situations, the disorder is not inherited from the parents.
Most cases of KCNQ2 developmental and epileptic encephalopathy occur de novo (caused by a spontaneous mutation and not inherited from a parent). However, a small number of patients affected by KCNQ2 inherited the mutated gene from a parent. In many of these cases, the parent may have few, if any, symptoms of the disease, in comparison to the child, because only some cells in the parent’s body contain a copy of the affected gene, in a condition known as mosaicism.
A parent who is mosaic for the KCNQ2 mutation may have enough properly functioning copies of the KCNQ2 gene (and thus enough correctly formed potassium channels) to exhibit no clinical symptoms, but the mutation may be passed to a child who then may carry the mutated gene in all of their cells.
Affected Populations
KCNQ2 equally affects males and females, and equally affects individuals across ethnic backgrounds. Cases often are undiagnosed or misdiagnosed, making it difficult to determine the disorder’s true frequency in the general population. In addition, the relatively recent discovery of this disorder means that older patients exist in the community who have not been tested or have been given another, incorrect, diagnosis.
Although KCNQ2-related disorders are rare, they have been frequently identified in studies that examine the effectiveness of genetic testing for individuals with suspected genetic epilepsies. In these studies, KCNQ2 consistently appears as one of the most commonly involved genes.
In a 2019 prospective cohort study in Scotland, KCNQ2 was by far the most common single-gene neonatal-onset epilepsy, with an estimated incidence of 1 per 17,000 live births. [Symonds et al 2019]